Species identification with DNA does not work well if your unknown search sequence is not represented in the DNA library. When we produce new sequences and check BOLD or Genbank for similar sequences, we sometimes obtain a “hit-list” of completely unrelated taxa. In the picture below, you see, for example, that the BOLD library has sequences of monkeys, flies, mites, and moths that are all about 70 % similar to the crustacean isopod that we tried to identify with the BOLD identification engine.
This and many other examples show that identification with DNA sequence similarity makes no sense if the target organism is not represented in the DNA library. This should also be a lesson for biodiversity studies based on environmental DNA (E-DNA).
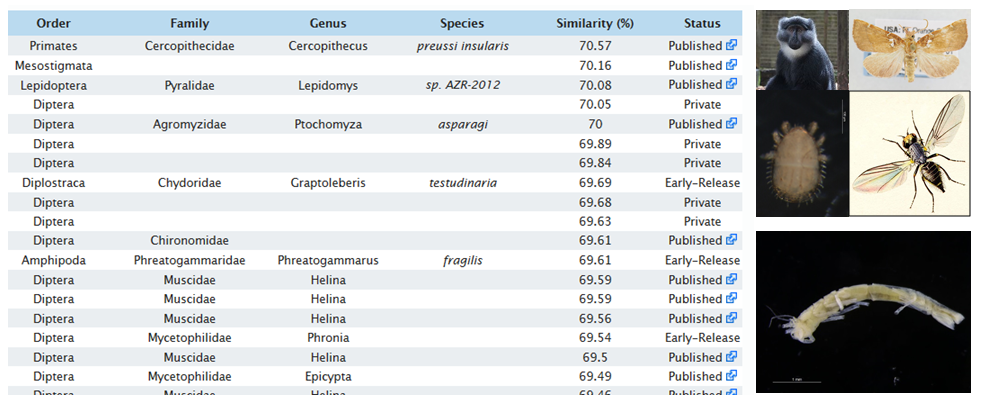
When sequences from an organism are not represented in the DNA-library, identification will fail. The search results show that there are many unrelated groups (pictures to right) that are 70 % similar to a crustacean isopod sequence (bottom right) .
It is well known that Genbank (and BOLD) contain many sequences with incorrect species name. This may have different reasons. Some of the problems can be traced to PCR-amplification error. To amplify a particular gene fragment (the barcode gene) with the polymerase chain reaction (PCR) we depend on a so-called primer. The primer is a short sequence (about 20 nucleotides long) that is supposed to find a complementary and relatively conserved stretch of DNA in the target sequence and be the starting point for the construction of a new complimentary DNA string.
Unfortunately, in some cases that does not happen because the priming site in the target organism is very different from the primer. There is apossibility then that the PCR may amplify DNA from another source instead. In animal samples preserved in alcohol there is usually DNA from other organisms as well; it may be remains of food, parasites, or even other larger animals (such as Homo sapiens) that have been in contact with the preserved specimen.
It seems likely that this is this is what happened when researchers thought they had sequenced crabs of the genera Ethusa and Philippidorippe and deposited their sequences in Genbank. They did not realize that their “crab” sequences actually are more or less identical with sequences from spiny lobsters. Thus the crab phylogeny that was published from these contaminated sequences is clearly problematic. We have also tried to make barcodes for Ethusa species in our projects, but without success, so we suspect that this is the explanation for the error in Genbank.
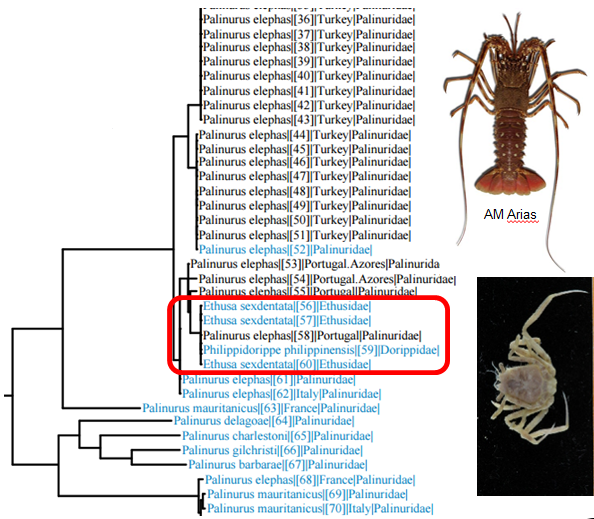
BOLD search with crab sequences from Genbank shows that the sequences are actually from spiny lobsters.
In other cases we find sequences in the DNA library that are very similar, but that carry different species names.
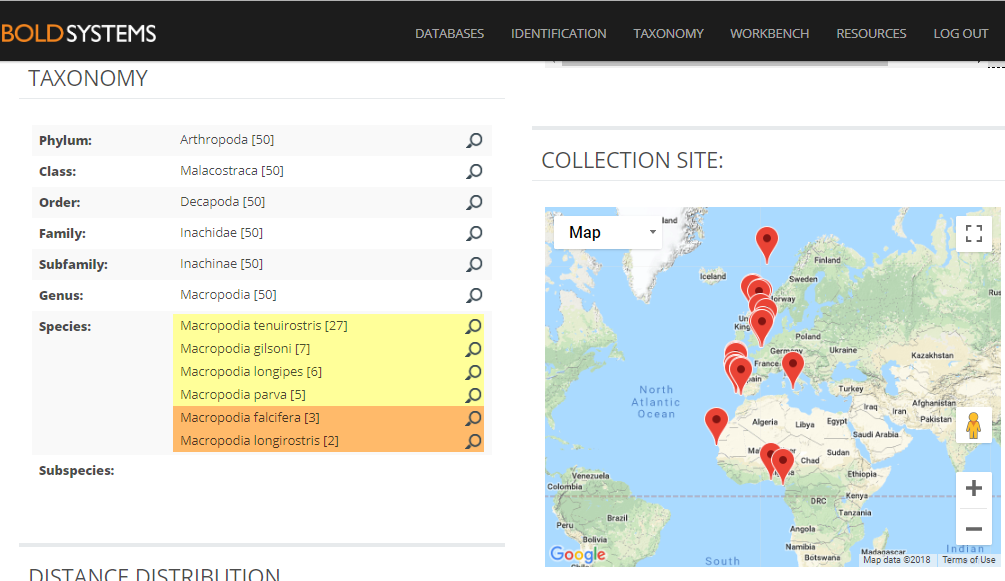
A collection of very similar sequences from spider crabs collected from South Africa to the Norwegian Sea have been identified as seven different species by different identifiers in different research labs.
It should not come as a surprize that such data will be reflected by the identification engine.

Identification search in BOLD with a spider crab sequence that returns seven possible species names.
The identification engine says: “A species level match could not be made. The queried specimen is likely to be one of the following species.” Cases like the one above clearly call for taxonomic evaluation. Are these sequences actually misidentified by the people that deposited them in the sequence library? Or, are these seven taxa actually representing “good species” that cannot really be discriminated with this barcode marker? Perhaps they diverged relatively recently, but did nor change much in their mitochondrial DNA? Alternatively, are the seven different species names an example of taxonomic oversplitting? It may happen when scientific subcultures in different parts of the world do not have a sufficient global perspective on their regional fauna. So is it really acceptable to consider Macropodia falcifera as an endemic species in South Africa, as stated in Wikipedia?
This exemplifies yet another advantage of DNA-barcoding. It can be an eye-opener for taxonomic problems of different sorts that require more studies to be resolved.
Another striking consequence of the DNA barcoding campaign is that many species are much more genetically diverse than expected. Our work in the museum has revealed several cases of species with up to 30 % genetic difference in the mitochondrial barcode sequence. Such observations suggest that we may be dealing with undescribed “cryptic species” and demonstrates how DNA-barcoding has also become an important instrument in species discovery.
The issues I have described in this blog post need to be addressed in a so-called “taxonomic feed-back loop” with emended information to the DNA-barcode library. Only then can we hope to get our understanding of the units of biodiversity right and hope that we can correctly identify them with barcodes.
– Endre
PS: Make sure to catch the previous posts on DNA barcoding in our advent calendar as well, you can find them here:
Door #5: DNA-barcoding with BOLD